


Neuroinflamación:
Resumen
Múltiples líneas de evidencia apoyan el papel patogénico de la neuroinflamación en enfermedades psiquiátricas. Si bien las enfermedades autoinmunes sistémicas son causas bien documentadas de trastornos neuropsiquiátricos, las encefalitis autoinmunes sinápticas con síntomas psicóticos a menudo son menos reconocidas. Paralelo al vínculo entre los síntomas psiquiátricos y la autoinmunidad en las enfermedades autoinmunes, se producen anomalías neuroinmunológicas en los trastornos psiquiátricos clásicos (por ejemplo, trastorno depresivo mayor, trastorno bipolar, esquizofrenia y trastorno obsesivo-compulsivo). Las investigaciones sobre la fisiopatología de estas afecciones tradicionalmente hacían hincapié en la desregulación de los sistemas glutamatérgico y monoaminérgico, pero los mecanismos que causaban estas anomalías neurotransmisoras seguían siendo difíciles de alcanzar. Revisamos el vínculo entre la autoinmunidad y los trastornos neuropsiquiátricos, y la evidencia humana y experimental que respalda el papel patogénico de la neuroinflamación en los trastornos psiquiátricos clásicos seleccionados. Comprender cómo interactúan los sistemas psicosociales, genéticos, inmunológicos y de neurotransmisores puede revelar pistas patogénicas y ayudar a identificar nuevas terapias preventivas y sintomáticas.
Palabras clave:
- Neuroinflamación,
- Psychoneuroinmunology,
- Astrocito,
- Microglia,
- Cytokines,
- Estrés oxidativo
- Depresión,
- Desorden obsesivo compulsivo,
- Trastorno bipolar, esquizofrenia
Introducción
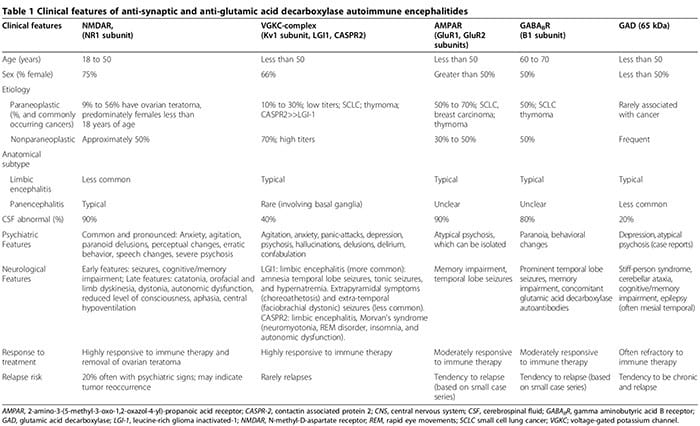
A medida que las anomalías biológicas se identifican cada vez más entre los pacientes con trastornos psiquiátricos, la distinción entre enfermedad neurológica y psiquiátrica se desvanece. Además de las enfermedades autoinmunes sistémicas asociadas con manifestaciones psiquiátricas (por ejemplo, lupus) [1], más recientemente, se identificaron pacientes con psicosis aguda aislada con encefalitis autoinmune sináptica (Tabla 1) [2-6]. A estos pacientes a menudo se les diagnostica erróneamente trastornos psicóticos primarios refractarios, lo que retrasa el inicio de una terapia inmune efectiva (Tabla 1). Además, la creciente evidencia respalda el papel patogénico de los anticuerpos antineuronales en los trastornos neuropsiquiátricos [7].
La separación de los trastornos neurológicos y psiquiátricos, respaldada por la concepción de Descartes de la 'mente' como una entidad ontológicamente distinta y por la reproducibilidad de las anomalías neuropatológicas, dominó la medicina en 19th y principios de 20th siglos [8]. Desde entonces, una colección en expansión de causas biológicas reproducibles, desde neurosífilis, traumatismo craneoencefálico, apoplejía, tumor, desmielinización y muchos otros causó complejos de síntomas que se superponen con los trastornos psiquiátricos clásicos [9-11]. Más recientemente, se han documentado anomalías neuroinflamatorias e inmunológicas en pacientes con trastornos psiquiátricos clásicos.
Los moduladores inmunes periféricos pueden inducir síntomas psiquiátricos en modelos animales y humanos [12-19]. Los animales sanos inyectados con citocinas proinflamatorias IL-1β y factor de necrosis tumoral alfa (TNF-α) muestran un “comportamiento de enfermedad” asociado con el aislamiento social [12]. En humanos, las inyecciones de endotoxina en dosis bajas desactivan el cuerpo estriado ventral, una región crítica para el procesamiento de recompensas, lo que produce anhedonia, un síntoma depresivo debilitante [14]. Aproximadamente el 45% de los pacientes con cáncer y hepatitis C no deprimidos tratados con IFN-α desarrollan síntomas depresivos asociados con niveles séricos elevados de IL-6 [12,15,17,18].
Las condiciones médicas asociadas con anomalías inflamatorias e inmunológicas crónicas, como la obesidad, la diabetes, las neoplasias malignas, la artritis reumatoide y la esclerosis múltiple, son factores de riesgo para la depresión y el trastorno bipolar [10,12,13,15,17,18]. lo positivo la correlación entre estas condiciones médicas y la enfermedad psiquiátrica sugiere la presencia de un proceso inflamatorio subyacente generalizado que afecta el cerebro entre otros órganos [10,19,20]. Un estudio basado en la población de 30-year mostró que tener un enfermedad autoinmune o una hospitalización previa por infección grave aumentó el riesgo de desarrollar esquizofrenia por 29% y 60%, respectivamente [16]. Además, el virus del herpes simple, Toxoplasma gondii, citomegalovirus e influenza durante el embarazo aumentan el riesgo de desarrollar esquizofrenia [16].
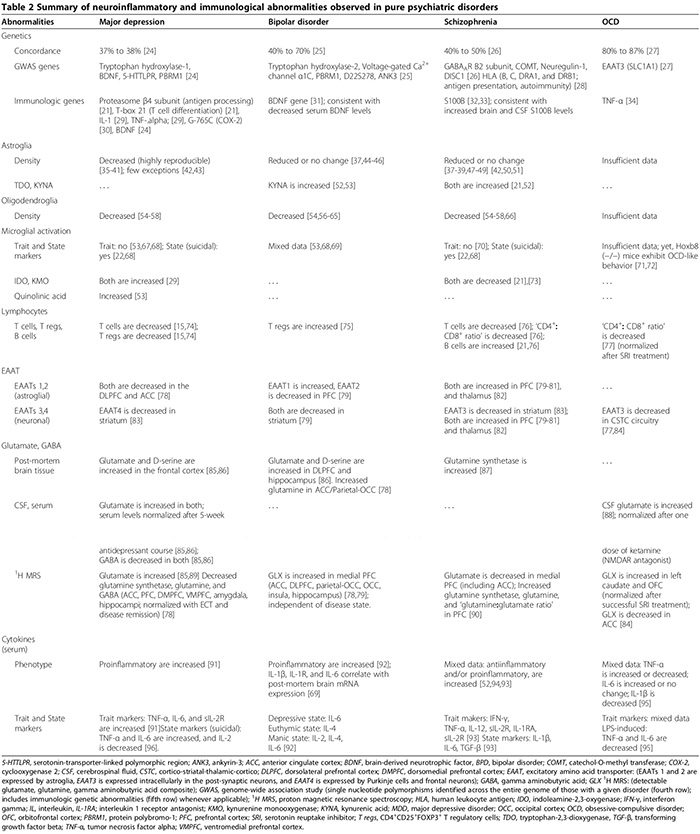
Las anomalías inmunológicas humorales y celulares periféricas [21,22] (Tabla 2) [13,21-23] son más frecuentes en pacientes psiquiátricos en relación con los controles sanos. Tanto en estudios piloto (n = 34 pacientes con trastorno depresivo mayor (MDD), n = 43 controles sanos) como en estudios de replicación (n = 36 MDD, n = 43 controles sanos), un análisis de suero que comprende nueve biomarcadores séricos distinguió a los sujetos con MDD de los sanos. controles con 91.7% de sensibilidad y 81.3% de especificidad; los biomarcadores significativamente elevados para los síntomas neuropsiquiátricos fueron las moléculas inmunológicas alfa 1 antitripsina, mieloperoxidasa y el receptor soluble TNF-α II [23].
 Primero revisamos la asociación entre autoinmunidad y trastornos neuropsiquiátricos, que incluyen: 1) lupus eritematoso sistémico (LES) como un prototipo de enfermedad autoinmune sistémica; 2) encefalitis autoinmunes asociadas con autoanticuerpos séricos anti-sináptica y ácido glutámico descarboxilasa (GAD); y 3) trastornos autoinmunitarios neuropsiquiátricos pediátricos asociados con infecciones estreptocócicas (PANDAS) y trastorno obsesivo-compulsivo puro (TOC) asociados con autoanticuerpos anti- basales / talámicos. A continuación, discutimos el papel de la inflamación / autoinmunidad innata en los trastornos psiquiátricos clásicos, incluidos el TDM, el trastorno bipolar (DBP), la esquizofrenia y el TOC.
Primero revisamos la asociación entre autoinmunidad y trastornos neuropsiquiátricos, que incluyen: 1) lupus eritematoso sistémico (LES) como un prototipo de enfermedad autoinmune sistémica; 2) encefalitis autoinmunes asociadas con autoanticuerpos séricos anti-sináptica y ácido glutámico descarboxilasa (GAD); y 3) trastornos autoinmunitarios neuropsiquiátricos pediátricos asociados con infecciones estreptocócicas (PANDAS) y trastorno obsesivo-compulsivo puro (TOC) asociados con autoanticuerpos anti- basales / talámicos. A continuación, discutimos el papel de la inflamación / autoinmunidad innata en los trastornos psiquiátricos clásicos, incluidos el TDM, el trastorno bipolar (DBP), la esquizofrenia y el TOC.
Trastornos neuropsiquiátricos asociados con autoinmunidad
Lupus eritematoso sistémico
Entre el 25 % y el 75 % de los pacientes con LES tienen afectación del sistema nervioso central (SNC), y los síntomas psiquiátricos suelen presentarse dentro de los primeros dos años del inicio de la enfermedad. Los síntomas psiquiátricos pueden incluir ansiedad, estado de ánimo y trastornos psicóticos [97]. La resonancia magnética (RM) cerebral es normal en aproximadamente el 42% de los casos de LES neuropsiquiátrico [97]. La microangiopatía y la ruptura de la barrera hematoencefálica (BBB) pueden permitir la entrada de autoanticuerpos en el cerebro [97]. Estos anticuerpos incluyen anti-ribosomal P (positivo en el 90% de los pacientes con LES psicóticos) [1], anti-células endoteliales, anti-gangliósido, anti-dsDNA, subunidades anti-2A/2B de los receptores de N-metil-D-aspartato ( NMDAR) y anticuerpos antifosfolípidos [97]. Citocinas proinflamatorias, principalmente IL-6 [97], S100B [97], la molécula de adhesión intracelular 1 [97] y la metaloproteinasa de matriz-9 [98] también están elevadas en el LES. Recientemente se revisaron las manifestaciones psiquiátricas del LES, la enfermedad de Sjögren, el síndrome de Susac, la vasculitis del SNC, la enfermedad de Whipple del SNC y la enfermedad de Behçet [1].
Encefalitis neuropsiquiátricas autoinmunes asociadas con antisináptico y descarboxilasa del ácido glutámico en suero
Autoanticuerpos
Las encefalitis autoinmunes se caracterizan por un inicio agudo de convulsiones del lóbulo temporal, características psiquiátricas y déficits cognitivos [2,3,99-108]. La fisiopatología suele estar mediada por autoanticuerpos que se dirigen a autoantígenos sinápticos o intracelulares en asociación con un origen paraneoplásico o no paraneoplásico [3]. Los autoanticuerpos antisinápticos se dirigen a las subunidades NR1 de NMDAR [100,108,109], complejos de canales de potasio dependientes de voltaje (VGKC) (subunidad Kv1, glioma rico en leucina inactivado (LGI1) y proteína 2 asociada a la contactina (CASPR2)) [101,102,106], GluR1 y subunidades GluR2 del receptor del ácido amino-3-hidroxi-5-metil-4-6,110,111-isoxazolpropiónico (AMPAR) [1] y subunidades B3,99,103 de los receptores B del ácido γ-aminobutírico (GABABR) [65]. Los autoanticuerpos antiintracelulares se dirigen a los autoantígenos onconeuronales y GAD-2,3 [XNUMX].
La inflamación asociada con autoanticuerpos anti-sinápticos, particularmente autoanticuerpos NMDAR, es típicamente mucho más leve que la asociada con autoanticuerpos GAD o autoanticuerpos antineuronales relacionados con trastornos autoinmunes sistémicos o síndromes paraneoplásicos [2,107].
Aunque finalmente surgen síntomas neurológicos, las manifestaciones psiquiátricas, que van desde la ansiedad [2,3] hasta la psicosis que imita la esquizofrenia [2-6], pueden inicialmente dominar o preceder a las características neurológicas. Hasta dos tercios de los pacientes con encefalitis autoinmune anti-NMDAR, inicialmente presentes en los servicios psiquiátricos [5]. Las encefalitis autoinmunes mediadas por anticuerpos anti-sinápticos deben ser consideradas en el diferencial de la psicosis aguda [2-6]. Las presentaciones psiquiátricas pueden incluir resonancia magnética cerebral normal y análisis de líquido cefalorraquídeo (LCR), sin encefalopatía ni convulsiones [2,3,5,6,107]. Reportamos un caso de autoanticuerpos GAD seropositivos asociados con neuroinflamación comprobada por biopsia, a pesar de los análisis normales de MRI cerebral y CSF, donde el paciente presentaba psicosis aislada diagnosticada como esquizofrenia según los criterios del Manual Diagnóstico y Estadístico de Trastornos Mentales, edición 4 (DSM-IV) [2]. Además, las encefalitis autoinmunes seronegativas también pueden presentar alteraciones neuropsiquiátricas prominentes, lo que hace que el diagnóstico sea más elusivo [107,112,113]. Las características psiquiátricas y neurológicas asociadas con autoanticuerpos anti-sinápticos y GAD se resumen en la Tabla 1 [1-6,99-108,114].
Los autoanticuerpos séricos antisinápticos y GAD pueden aparecer en pacientes con trastornos psiquiátricos puros [2,4,5,112,115-121]. En una cohorte prospectiva de 29 sujetos que cumplieron con los criterios del DSM-IV para la esquizofrenia, se encontraron autoanticuerpos séricos anti-NMDAR en tres sujetos y autoanticuerpos anti-VGKC-complejo en un sujeto [5]. Utilizando técnicas más sensibles para detectar autoanticuerpos de inmunoglobulina G (IgG) NR1 en 100 pacientes con esquizofrenia definitiva, no se identificaron autoanticuerpos [122]. Sin embargo, este estudio no evaluó los autoanticuerpos dirigidos a la subunidad NR2 de NMDAR. Otros estudios informaron un aumento significativo de las probabilidades de niveles elevados de anticuerpos NR90 (niveles de control no psiquiátricos del ≥ percentil 2) (odds ratio (OR) 2.78, intervalo de confianza (IC) del 95%: 1.26 a 6.14, P = 0.012) entre individuos con manía (n = 43), pero no en manía crónica o esquizofrenia [116].
PANDAS y trastorno obsesivo-compulsivo puro asociado con autoanticuerpos anti-ganglios basales / talámicos
El TOC a menudo complica los trastornos neurológicos que afectan a los ganglios basales, como la corea de Sydenham, la enfermedad de Huntington y la enfermedad de Parkinson. Los anticuerpos contra los ganglios basales están implicados en la corea de Sydenham [123]. PANDAS se caracteriza por exacerbaciones agudas de los síntomas del TOC y/o tics motores/fónicos después de una infección prodrómica por estreptococos β-hemolíticos del grupo A. Se cree que la fisiopatología implica una reactividad cruzada entre los anticuerpos antiestreptocócicos y las proteínas de los ganglios basales [124]. La superposición clínica entre los PANDAS y el TOC puro sugiere un mecanismo etiológico común [125].
Entre una cohorte aleatoria de 21 pacientes con TOC puro, el 91.3 % tenía anticuerpos contra los ganglios basales del LCR (P <0.05) y autoanticuerpos antitalámicos (P <0.005) a 43 kDa [88], en paralelo con las anomalías funcionales en el sistema corticoestriado-tálamo. -circuito cortico de sujetos con TOC [84]. Otro estudio documentó que el 42 % (n = 21) de los sujetos pediátricos y adolescentes con TOC tenían autoanticuerpos séricos contra los ganglios basales a 40, 45 y 60 kDa en comparación con el 2 % al 10 % de los controles (P = 0.001) [7]. Se detectaron autoanticuerpos anti-ganglios basales en los sueros del 64 % de los sujetos PANDAS (n = 14), en comparación con solo el 9 % (n = 2) de los controles positivos para estreptococos/negativos para TOC (P <0.001) [126] . Un estudio no encontró diferencias entre la prevalencia de autoanticuerpos contra los ganglios basales en el TOC (5.4 %, n = 4) frente a los controles con TDM (0 %) [127]; sin embargo, una limitación fue el uso aleatorio de corteza de rata y ganglios basales y corteza de bovino que podría haber limitado la identificación de casos seropositivos.
Los autoantígenos de los ganglios basales son la aldolasa C (40 kDa), la enolasa neuronal específica/no neuronal (doblete de 45 kDa) y la piruvato quinasa M1 (60 kDa), enzimas glucolíticas neuronales, que participan en la neurotransmisión y el metabolismo neuronal.
Página 3 de 24 y señalización celular [128]. Estas enzimas muestran una homología estructural sustancial con las proteínas estreptocócicas [129]. El último estudio (96 OCD, 33 MDD, sujetos con esquizofrenia 17) probó el suero del paciente contra piruvato quinasa, aldolasa C y enolasa, específicamente; una mayor proporción de sujetos con TOC fueron seropositivos con respecto a los controles (19.8% (n = 19) frente a 4% [n = 2], P = 0.012) [130].
Sin embargo, en el mismo estudio solo uno de los sujetos con TOC seropositivo 19 también tenía anticuerpos anti-estreptolisina O positivos, lo que sugiere que en el TOC puro, la seronegatividad del anticuerpo anti-estreptolisina O no excluye la presencia de autoanticuerpos anti-ganglios basales. .
En el TOC puro, la seropositividad para los ganglios anti-basales / anticuerpos talámicos se asocia con mayores niveles de glicina CSF (P = 0.03) [88], lo que sugiere que estos anticuerpos contribuyen a la hiperglutamatérgia observada en el TOC [84,88,131]. La mejora del TOC provocado por infección con terapias inmunológicas apoya la patogenicidad de estos autoanticuerpos [132]. Se está llevando a cabo un gran ensayo NIH que evalúa la eficacia de la inmunoglobulina intravenosa (IGIV) en niños con TOC de inicio agudo y anticuerpos antiestreptococos (ClinicalTrials.gov: NCT01281969). Sin embargo, el hallazgo de niveles ligeramente más altos de glutamato en CSF en pacientes con TOC con CSF negativos anti-ganglios básales / talámicos en comparación con aquellos con anticuerpos CSF positivos sugiere que los mecanismos no inmunológicos pueden desempeñar un papel en el TOC [84]. Otros mecanismos, incluida la inflamación mediada por citocinas (Tabla 2), también se hipotetizan.
Trastornos psiquiátricos asociados con inflamación innata
Los trastornos de inflamación/autoinmunidad innata ocurren en algunos pacientes con trastornos psiquiátricos clásicos. Discutimos las anormalidades del SNC relacionadas con la inflamación innata, que incluyen patología glial, niveles elevados de citocinas, activación de ciclooxigenasa, desregulación del glutamato, niveles aumentados de S100B, aumento del estrés oxidativo y disfunción BBB, en MDD, BPD, esquizofrenia y TOC. También describimos cómo la inflamación innata puede vincularse mecánicamente con las anomalías monoaminérgicas y glutamatérgicas tradicionales informadas en estos trastornos (Figuras 1 y 2). También se revisa el papel terapéutico de los agentes antiinflamatorios en los trastornos psiquiátricos.
 Histopatología astroglial y oligodendroglial
Histopatología astroglial y oligodendroglial
 Histopatología astroglial y oligodendroglial
Histopatología astroglial y oligodendroglialAstroglia y oligodendroglia son esenciales para los nervios metabólico homeostasis, comportamiento y funciones cognitivas superiores [54-56,133-136]. La astroglía inactiva normal proporciona energía y apoyo trófico a las neuronas, regula la neurotransmisión sináptica (Figura 2), la sinaptogénesis, el flujo sanguíneo cerebral y mantiene la integridad de la BHE [134,136,137]. La oligodendroglia madura proporciona energía y apoyo trófico a las neuronas, mantiene la integridad de la BBB y regula la reparación axonal. y mielinización de tractos de sustancia blanca que proporcionan conectividad inter e intrahemisférica [54-56]. Tanto la astroglia como la oligodendroglia producen citoquinas antiinflamatorias que pueden regular negativamente la inflamación nociva [52,55].
En MDD, la pérdida astroglial es un hallazgo consistente post mortem en áreas funcionalmente relevantes, incluyendo la corteza cingulada anterior, la corteza prefrontal, la amígdala y la sustancia blanca [35-38,42-46,55,138-147], con algunas excepciones [42,43]. Los estudios post-mortem revelaron una reducción de la densidad astrogliada de la proteína ácida fibrilar glial (GFAP), principalmente en la corteza prefrontal [37,38] y la amígdala [36]. Un gran análisis proteómico de cortezas frontales de pacientes deprimidos mostró reducciones significativas en tres isoformas GFAP [39]. Aunque en un estudio que informó que no había pérdida glial significativa, el análisis de subgrupos reveló una disminución significativa (75%) en la densidad astrogliada GFAP-positiva entre sujetos de estudio menores de 45 años de edad [35]. Un estudio morfométrico mostró similarmente que no hubo cambios en la densidad glial en cerebros MDD de vida tardía [148]. Presumimos que la aparente ausencia de pérdida astroglial entre los pacientes mayores con TDM puede reflejar una astrogliosis secundaria [35] que se asocia con una edad más avanzada [42,50] en lugar de una verdadera negativa.
Los estudios en animales son consistentes con los estudios en humanos que muestran pérdida astroglial en MDD. Las ratas Wistar-Kyoto, conocidas por exhibir comportamientos depresivos, revelaron una densidad astroglial reducida en las mismas áreas observadas en humanos [40]. La administración del agente tóxico para la astroglia, el ácido L-alfa-aminoadípico, induce síntomas de tipo depresivo en ratas, lo que sugiere que la pérdida de la astroglia es patógena en el TDM [41].
Los estudios post-mortem de sujetos con TDM documentaron una reducción de la densidad oligodendroglial en la corteza prefrontal y la amígdala [54-57,66], que puede correlacionarse con cambios cerebrales en la sustancia blanca focal observados ocasionalmente en algunos pacientes con TDM [57]. Sin embargo, las anomalías microvasculares también pueden contribuir a estos cambios [57].
En BPD, algunos estudios demuestran una pérdida glial significativa [138,143,149,150], mientras que otros no lo hacen [37,44-46]. Estos resultados inconsistentes pueden resultar de la falta de control para: 1) tratamiento con estabilizadores del estado de ánimo, debido a que el análisis post-hoc informado por algunos estudios mostró una reducción significativa en la pérdida glial solo después de controlar el tratamiento con ácido valproico y litio [46]; 2) formas familiares de DBP, ya que la pérdida glial es particularmente prominente entre los pacientes con TLP con un historial familiar fuerte [143]; y / o, 3) el estado predominante de depresión versus manía, ya que la pérdida glial es frecuente en MDD [35-38,42-46,55,138-147]. Si la astroglia o la oligodendroglia representan la mayoría de las pérdidas gliales no está clara; mientras que el análisis proteómico reveló una disminución significativa en una isoforma GFAP astroglial [39], varios otros estudios post-mortem encontraron sin cambios [36,37] o expresión astrogliada GFAP-positiva reducida en la corteza orbitrofrontal [47], o densidad oligodendroglial reducida [54- 56,58,59].
En la esquizofrenia, la pérdida astroglial es un hallazgo inconsistente [48,150]. Mientras que algunos estudios no mostraron una pérdida astroglia significativa [42,50,51], otros encontraron una densidad astrogliada reducida [37,38,43,44,48,49,151] y reducciones significativas en dos isoformas de GFAP [39]. Los hallazgos inconsistentes pueden ser el resultado de: 1) comorbilidad MDD, que a menudo se asocia con pérdida glial; 2) variación de la edad, ya que los pacientes mayores han aumentado la astroglia GFAP-positiva [35,42,50]; 3) regional [150] y variabilidad de la capa cortical [48]; 4) tratamiento con fármacos antipsicóticos, ya que los estudios experimentales muestran una reducción de [152] y un aumento [153] de la densidad astroglial relacionada con el tratamiento antipsicótico crónico [70]; y 5) estado de enfermedad (por ejemplo, comportamiento suicida versus no suicida) [154]. Los estudios post-mortem documentaron la pérdida oligodendroglial [54,56,60-65,148,155,156], particularmente en la corteza prefrontal, la corteza cingulada anterior y el hipocampo [148]. El examen ultraestructural de la región prefrontal mostró fibras anormalmente mielinizadas en materia gris y blanca; tanto la edad como la duración de la enfermedad se correlacionaron positivamente con las anomalías de la sustancia blanca [157].
En contraste con los trastornos neurodegenerativos que comúnmente se asocian con la proliferación astroglial [136], los trastornos psiquiátricos se asocian con una densidad astrogliada reducida o sin cambios [138]. La falta de densidad glial aumentada en los trastornos psiquiátricos de inicio temprano [44,138] puede reflejar la tasa más lenta de progresión degenerativa en enfermedades psiquiátricas [138].
Postulamos que los cambios degenerativos asociados con los trastornos psiquiátricos son más sutiles y no lo suficientemente graves como para provocar factores de transcripción intracelulares astrogliales que regulan positivamente la astrogliosis, incluido el activador del transductor de señal de la transcripción 3 y el factor nuclear kappa B (NF-κB) [136].
Si bien la mayoría de los estudios post-mortem se centraron en la alteración de la densidad glial en MDD, BPD y esquizofrenia, otros describieron la alteración de la morfología de las células gliales, con hallazgos mixtos. En MDD y BPD, el tamaño glial se aumenta o no cambia [55]. Un estudio encontró una reducción del tamaño de la glía en el TLP y la esquizofrenia, pero no en el TDM [43]. Un estudio post-mortem de pacientes deprimidos que se suicidaron encontró un aumento del tamaño astroglial en la sustancia blanca cingulada anterior, pero no en la corteza [158]. Un estudio en sujetos esquizofrénicos encontró un tamaño astroglial marcadamente disminuido en la capa V de la corteza prefrontal dorsolateral, a pesar de que la densidad astroglial es el doble que la de los controles en la misma capa [48]. Los resultados mixtos pueden reflejar parcialmente estudios anteriores de alteraciones gliales en enfermedades psiquiátricas que no especificaron astroglia versus oligodendroglia [148].
La pérdida glial en enfermedades psiquiátricas puede contribuir a la neuroinflamación a través de varios mecanismos, que incluyen niveles anormales de citoquinas (consulte la sección Citoquinas), metabolismo desregulado del glutamato (consulte la sección Glutamato), proteína S100B elevada (consulte la sección S100B), y una función alterada de la BHE (consulte la sección de la barrera Hematoencefálica), que da como resultado una cognición y un comportamiento alterados [44,45,54,133,159].
Histopatología microglial
Microglia son las células inmunes residentes del SNC. Brindan vigilancia inmune continua y regulan la poda sináptica del desarrollo [160,161]. La lesión del SNC transforma la microglía en reposo ramificada en células ameboides fagocíticas en forma de bastón alargadas y activadas que proliferan y migran hacia el sitio de la lesión a lo largo de gradientes quimiotácticos (es decir, activación y proliferación microglial (MAP)) [161]. Las células microgliales humanas expresan NMDAR que pueden mediar MAP que conduce a una lesión neuronal [162].
En MDD, BPD y esquizofrenia, los resultados de los estudios post-mortem que investigan la presencia de MAP son mixtos. Los estudios post-mortem revelaron un MAP elevado en solo uno de cada cinco sujetos con MDD [67]. En algunos pacientes con trastorno de TLP, se documentó un aumento de la microglia de leucocitos humanos positivos al DR con procesos engrosados en la corteza frontal [69]. En la esquizofrenia, mientras que algunos estudios informaron un MAP elevado en relación con los controles, otros no mostraron diferencias entre los grupos [22,67,70]. En un estudio post-mortem que evalúa MAP en MDD y BPD; la densidad de células microgliales positivas al ácido quinolínico se incrementó en la corteza cingulada anterior subgenual y en la corteza anterior mediada por la corteza de los pacientes con TDM y TLP que se suicidaron en relación con los controles [53]. El análisis post-hoc reveló que este MAP incrementado fue exclusivamente atribuible a MDD y no a BPD, ya que la tinción inmunológica microglial positiva en sujetos con MDD fue significativamente mayor que en el subgrupo BPD en las cortezas subgénero cingulado anterior y midcingulate, y desde la densidad de microglia fue similar en ambos grupos de BPD y control [53]. Un estudio que comparó los tres trastornos (nueve MDD, cinco BPD, catorce esquizofrenia, diez controles sanos) no demostró diferencias significativas en la densidad microglial en los cuatro grupos [68].
Estos resultados mixtos pueden atribuirse a marcadores inmunológicos microgliales variables utilizados entre diferentes estudios [70] y / o la falta de control para la gravedad de la enfermedad [22,53,68]. Notablemente, tres estudios post-mortem de MDD y sujetos esquizofrénicos documentaron una fuerte correlación positiva entre MAP y suicidalidad en la corteza cingulada anterior y el tálamo mediodorsal, independientemente del diagnóstico psiquiátrico [22,53,68]. Por lo tanto, MAP puede ser un marcador de estado en lugar de un rasgo para MDD y esquizofrenia.
En el TOC, los modelos animales sugieren que la disfunción y la reducción de ciertos fenotipos microgliales, como los que expresan el gen Hoxb8, que codifica el factor de transcripción de la homeobox, pueden causar un comportamiento similar al TOC [71,72].
Los ratones Hoxb8 knockout presentan un comportamiento de aseo y ansiedad excesivos en asociación con una densidad microglial reducida [71,72]. Este comportamiento de aseo excesivo se asemeja a las características conductuales del TOC humano. La inyección de Hoxb8 en ratones knockout Hoxb8 adultos revierte la pérdida microglial y restablece el comportamiento normal [71,72]. El papel de estos fenotipos microgliales específicos en el TOC humano no está claro.
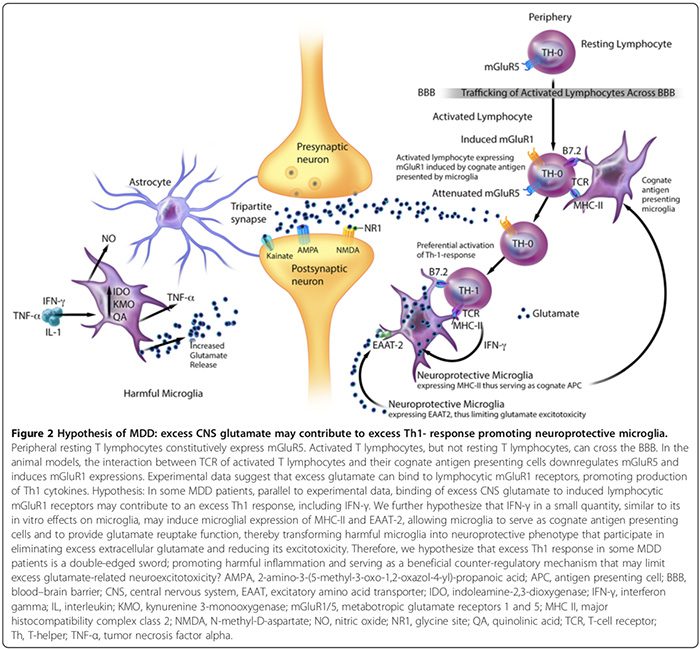
Los datos experimentales sugieren que MAP comprende fenotipos distintivos dañinos y neuroprotectores (Figura 2). La microglía dañina no expresa el complejo mayor de histocompatibilidad II (MHC-II) y, por lo tanto, no puede actuar como células presentadoras de antígeno (APC) [163,164]; promueven los efectos nocivos [17,69,165] a través de la producción de citocinas proinflamatorias, señalización de óxido nítrico sintasa [17,166], promoviendo la expresión glial y BBB-pericito / ciclooxigenasa endotelial-2 (COX-2) [167], induciendo la secreción de S100B astroglial (consulte la sección S100B) y liberación de glutamato microglial [17,136,168,169]. Microglia dañina también secreta prostaglandina E-2 (PGE-2) que promueve la producción de citoquinas proinflamatorias, que a su vez aumenta los niveles de PGE-2 en un ciclo de alimentación hacia delante [29]. Además, PGE-2 estimula la expresión COX-2, que media la conversión de ácido araquidónico a PGE-2, configurando otro ciclo de alimentación hacia adelante [29].
La microglia neuroprotectora por contraste puede: 1) expresar MHC-II in vivo e in vitro [163,166] y actuar como APC análogo (Figura 2) [163,164,166]; 2) facilitan la cicatrización y limitan la lesión neuronal promoviendo la secreción de citoquinas antiinflamatorias [17], factor neurotrófico derivado del cerebro [17] y factor de crecimiento similar a la insulina-1 [166]; y 3) expresan transportador de aminoácidos excitadores-2 (EAAT2) que elimina el exceso de glutamato extracelular [163,166] y promueve la autoinmunidad linfocítica T neuroprotectora (Figura 2) [163,164]. Sin embargo, se necesitan más estudios para confirmar el rol contribuyente de la microglía neuroprotectora a los trastornos neuropsiquiátricos en humanos.
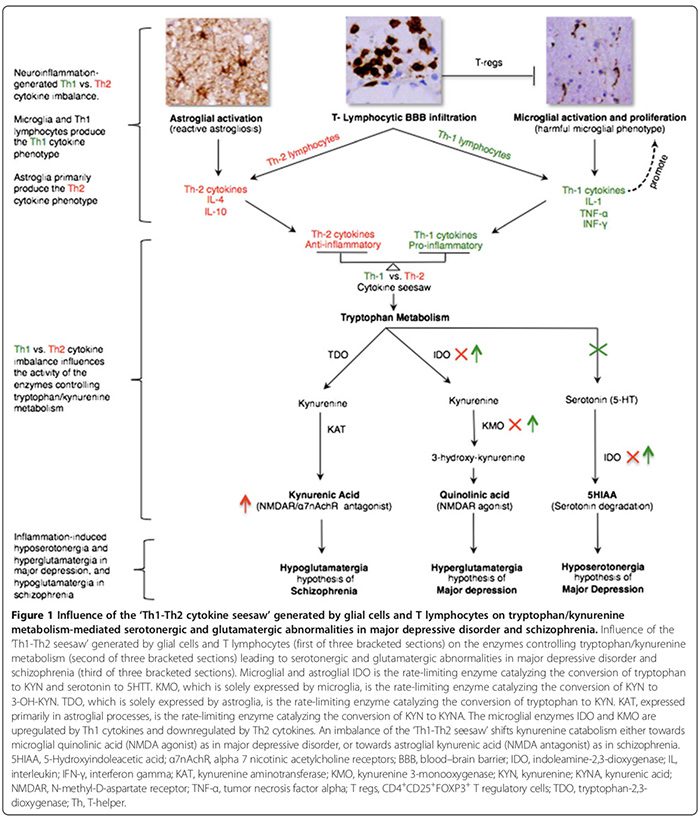
In vitro los estudios en animales sugieren que la proporción de microglía dañina versus neuroprotectora puede verse influenciada por el efecto neto de los mecanismos contrarreguladores inflamatorios [15,74,164,166]. Estos mecanismos incluyen el número de células T reguladoras neuroprotectoras CD4+CD25+FOXP3+ ((T regs) Figura 1) [15,74,164,166] y los niveles de citoquinas en el cerebro; los niveles bajos de IFN-γ pueden promover la microglía neuroprotectora (Figura 2) [166], mientras que los niveles altos pueden promover el fenotipo dañino [166].
El papel de las citoquinas
Las citoquinas proinflamatorias incluyen IL-1β, IL-2, IL-6, TNF-α e IFN-γ. Son secretados principalmente por microglía, linfocitos Th1 y monocitos/macrófagos de fenotipo M1 (Figura 1) [15,170]. Promueven la inflamación dañina. Las citocinas antiinflamatorias incluyen IL-4, IL-5 e IL-10. Son secretadas principalmente por astroglia, Linfocitos Th2, monotipos / macrófagos de fenotipo T regs y M2 [15,52,74]. Pueden limitar la inflamación nociva [15,74] convirtiendo el fenotipo MXNMX-pheno-proinflamatorio en el fenotipo MXNMXX-antiinflamatorio beneficioso [1], y potencialmente promoviendo el fenotipo microglial neuroprotector [2]. El papel de las citoquinas proinflamatorias / antiinflamatorias en los trastornos psiquiátricos está respaldado por varias líneas de evidencia (Figura 15, Tabla 15,17,74,163,166) [1].
En MDD, el metanálisis más reciente (29 estudios, 822 MDD, 726 controles sanos) de citocinas proinflamatorias séricas confirmó que los niveles del receptor soluble de IL-2, IL-6 y TNF-α aumentan en MDD (marcadores de rasgos) [ 91], mientras que IL-1β, IL-2, IL-4, IL-8 e IL-10 no son estadísticamente diferentes de los controles [91]. En un estudio primario de citoquinas que comparó los subgrupos de MDD (47 MDD suicidas, 17 MDD no suicidas, 16 controles de salud), tanto la IL-6 sérica como el TNF-α fueron significativamente más altos, mientras que los niveles de IL-2 fueron significativamente más bajos en sujetos con MDD que se suicidaron en relación con los otros dos grupos [96]. Este hallazgo sugiere que IL-6 y TNF-α también son marcadores de estado de MDD [96]. La disminución de los niveles séricos de IL-2 asociada con el comportamiento suicida agudo puede reflejar una mayor unión a su receptor regulado al alza en el cerebro; paralelo al metanálisis mencionado anteriormente que muestra un aumento del receptor de IL-2 soluble en MDD [91]. Los estudios que investigaron la importancia clínica de las citocinas en el TDM demostraron que los niveles séricos de citocinas se elevan durante los episodios depresivos agudos [171,172] y se normalizan después de un tratamiento exitoso, pero no fallido, con antidepresivos [17] y terapia electroconvulsiva [29]; estos hallazgos sugieren un posible papel patógeno de las citoquinas.
En la DBP, las alteraciones de las citocinas séricas se resumieron en una revisión reciente; TNF-α, IL-6 e IL-8 se elevan durante las fases maníaca y depresiva, mientras que IL-2, IL-4 e IL-6 se elevan durante la manía [92]. Otros estudios mostraron que los niveles de receptor de IL-1β e IL-1 en suero no son estadísticamente diferentes de los controles sanos [92], aunque los estudios de tejido documentaron niveles elevados de receptor de IL-1β e IL-1 en la corteza frontal de BPD [69].
En la esquizofrenia, los resultados de los estudios que investigan las anomalías de las citoquinas son contradictorios (Tabla 2). Mientras que algunos estudios encontraron tanto una disminución de las citocinas proinflamatorias séricas (IL-2, IFN-γ) como un aumento de las citocinas antiinflamatorias séricas y del LCR (IL-10) [52], otros encontraron citocinas séricas proinflamatorias y antiinflamatorias elevadas, con un predominio de tipo proinflamatorio [22,173,174 ]. Un metanálisis de citocinas (62 estudios, 2,298 esquizofrenia, 858 controles sanos) mostró niveles elevados de antagonista de IL-1R, sIL-2R e IL-6 [174]. Sin embargo, este estudio no tuvo en cuenta el uso de antipsicóticos, que se cree que aumentan la producción de citocinas proinflamatorias [52]. Un metanálisis de citocinas más reciente (40 estudios, 2,572 esquizofrénicos, 4,401 controles) que representaron los antipsicóticos, encontró que TNF-α, IFN-γ, IL-12 y sIL-2R están constantemente elevados en la esquizofrenia crónica independientemente de la actividad de la enfermedad (marcadores de rasgos), mientras que IL-1β, IL-6 y el factor de crecimiento transformante beta se correlaciona positivamente con la actividad de la enfermedad (marcadores de estado)[173]. Los cultivos celulares de células mononucleares de sangre periférica (PBMC) obtenidas de pacientes esquizofrénicos produjeron niveles más altos de IL-8 e IL-1β de forma espontánea y después de la estimulación con LPS, lo que sugiere un papel para los monocitos/macrófagos activados en la patología de la esquizofrenia [175].
En el TOC, los resultados de una encuesta aleatoria de citoquinas en suero y LCR, y estudios de PBMC estimulados con LPS, son inconsistentes [93-95,176-179]. Existe una correlación entre el TOC y un polimorfismo funcional en la región promotora del gen TNF-α [34], aunque los estudios de baja potencia no confirmaron esta asociación [180]. Por lo tanto, los resultados mixtos de los estudios que documentan niveles de citoquinas TNF-α aumentados o disminuidos [93,176-178] pueden reflejar su inclusión variable del subconjunto de sujetos con TOC con este polimorfismo particular en sus cohortes.
Polarización de la respuesta de citocinas en depresión mayor y esquizofrenia
Los fenotipos de respuesta de citocinas se clasifican como proinflamatorios Th1 (IL-2, IFN-γ) o antiinflamatorios Th2 (IL-4, IL-5, IL-10) según las funciones inmunitarias que regulan. Mientras que las citocinas Th1 regulan la inmunidad mediada por células dirigida contra antígenos intracelulares, las citocinas Th2 regulan la inmunidad humoral dirigida contra antígenos extracelulares [29,52]. Las citoquinas Th1 son producidas por linfocitos Th1 y monocitos M1, mientras que las citoquinas Th2 son producidas por linfocitos Th2 y monocitos M2 [29,52]. En el cerebro, la microglia secreta predominantemente citoquinas Th1, mientras que la astroglia secreta predominantemente citoquinas Th2 [29,52]. La relación recíproca de las citocinas Th1:Th2, en lo sucesivo "balancín Th1-Th2", está influenciada por la proporción de microglía activada (exceso de Th1) a astroglía (exceso de Th2) y la interacción entre las células T activadas y el exceso de glutamato en el SNC. niveles que, según nuestra hipótesis, favorecen la respuesta Th1 (Figura 2) [29,163,166].
El desequilibrio de balancín Th1-Th2 puede influir en el metabolismo del triptófano al alterar sus enzimas [21,52], cambiando el catabolismo del triptófano hacia la kinurenina (KYN) y el catabolismo KYN hacia cualquiera de sus dos metabolitos descendentes; ácido quinolínico microglia que es mediada por la respuesta de Th1 o ácido cinurénico astroglial (KYNA) (Figura 1) que es mediada por la respuesta de Th2 [21,29,170].
Las enzimas del metabolismo del triptófano afectadas por el balancín Th1-Th2 incluyen (Figura 1): indoleamina 2,3-dioxigenasa (IDO) expresada por microglia y astroglia, las enzimas limitantes de la velocidad que median la conversión de triptófano a KYN y serotonina a 5- ácido hidroxiindolacético [21,29]. La quinurenina 3-monooxigenasa (KMO), expresada únicamente por la microglía, es la enzima limitante de la velocidad que convierte KYN en 3-hidroxiquinurenina (3-OH-KYN), que luego se metaboliza en ácido quinolínico [21,29]. La triptófano-2,3-dioxigenasa (TDO), expresada únicamente por la astroglía, es la enzima limitante de la velocidad que convierte triptófano a KYN [21,29]. La quinurenina aminotransferasa (KAT), expresada principalmente en procesos astrogliales, es la enzima limitante de la velocidad que media la conversión de KYN en KYNA [21,29].
Las citocinas Th1 activan IDO y KMO microgliales, cambiando el catabolismo KYN microglial hacia quinolínico. síntesis de ácido (agonista de NMDAR), mientras que las citocinas Th2 activan el IDO y el KMO microgliales, desplazando el catabolismo de KYN astroglial hacia la síntesis de KYNA (antagonista de NMDAR) mediada por TDO y KAT (Figura 1) [21,29].
Se han propuesto inmunofenotipos predominantes Th1 y Th2 para MDD y esquizofrenia, respectivamente, basados en patrones de citocinas periféricas, en lugar de en el SNC [52,173]. Creemos que los patrones de citocinas periféricas son marcadores sustitutos poco fiables de los del SNC. De hecho, los niveles de citoquinas periféricas pueden verse influenciados por muchas variables extra-SNC, que no se controlan de manera consistente en varios de los estudios de citoquinas periféricas, que incluyen: 1) edad, índice de masa corporal, medicamentos psicotrópicos, tabaquismo, estrés y fluctuaciones circadianas; 2) la influencia de actividad/estado de la enfermedad en la producción de síntesis de citoquinas seleccionadas [95,173]; y 3) los efectos de los agentes psicotrópicos sobre la producción de citocinas [52]. Las vidas medias cortas y la rápida renovación de las citoquinas séricas [181] (por ejemplo, 18 minutos para TNF-α [182] versus 60 minutos para IL-10 [183]), pueden limitar aún más la confiabilidad de la interpretación. sus niveles medidos a partir de muestras de suero aleatorias.
En MDD, existe un consenso de que predomina una respuesta de inmunofenotipo Th1 proinflamatoria (Tabla 2) [17,29]. Los altos niveles de ácido quinolínico en cerebros con TDM post mortem [53] sugieren la presencia de una respuesta Th1 regulada al alza (Figura 1) [21,29]. El ácido quinolínico del SNC elevado puede promover la apoptosis mediada por la entrada de calcio de la astroglía humana [184], lo que hipotéticamente puede mitigar el respuesta de Th2 derivada de astroglia [29], inclinando Th1 versus balanceo de balancín Th2 a favor de la respuesta microglial de Th1. La hiposerotonergia del SNC [29] agrega más apoyo a un exceso de respuesta de Th1, que reduce la síntesis de serotonina en el SNC [185] y aumenta su degradación (Figura 1) [21,29].
La hiperglutamatergia del SNC también puede contribuir a un exceso de respuesta de Th1 en el cerebro (Figura 2). Un estudio in vitro sugiere que los linfocitos T en reposo periféricos expresan metabotrópicamente el receptor de glutamato 5 (mGluR5) [164], cuya unión al glutamato inhibe la liberación linfocítica de IL-6, regulando negativamente la proliferación de células T-efectoras autorreactivas [164]. Los linfocitos T activados, pero no los linfocitos T en reposo, pueden cruzar el BBB [37].
Los datos experimentales sugieren que la interacción entre los receptores de células T de los linfocitos T activados y sus células presentadoras de antígeno afines pueden regular a la baja mGluR5 e inducir expresiones de mGluR1 [164]. En modelos animales, la unión del exceso de glutamato a los receptores linfocíticos mGluR1 promueve la producción de citocinas Th1, incluido el IFN-γ [164].
Presumimos que en algunos pacientes con MDD, paralelamente a los datos experimentales [164], la unión del exceso de glutamato del SNC a los receptores mGluR1 linfocíticos inducidos puede contribuir a una respuesta Th1 excesiva, incluido el IFN-γ (Figura 2). Especulamos que IFN-γ en una pequeña cantidad, similar a sus efectos in vitro sobre la microglía [166], puede inducir la expresión microglial de MHC-II y EAAT2 [163,166], lo que permite que la microglía sirva como células presentadoras de antígeno afines y proporcione glutamato. función de recaptación [163,164,166], transformando así la microglía dañina en un fenotipo neuroprotector [163,166] que participa en la eliminación del exceso de glutamato extracelular [163,164,166]. Por lo tanto, también planteamos la hipótesis de que el exceso de respuesta Th1 en subgrupos de pacientes con MDD es un arma de doble filo, que promueve una inflamación dañina y actúa como un mecanismo contrarregulador beneficioso que puede limitar el exceso de neuroexcitotoxicidad relacionada con el glutamato (Figura 2).
En la esquizofrenia, mientras que algunos estudios de citoquinas periféricas sugieren el predominio de un inmunofenotipo/respuesta antiinflamatoria Th2 [52], otros lo refutan [173,174]. Sin embargo, estamos de acuerdo con los autores que plantearon la hipótesis de que la respuesta Th2 es el fenotipo dominante en la esquizofrenia [52]. Los niveles elevados de KYNA en el cerebro, el LCR y el suero [21,52] sugieren una regulación a la baja de IDO y KMO microglial, que es una función de la respuesta Th2 que cambia el catabolismo de KYN astroglial hacia la síntesis de KYNA (Figura 1) [21,52]. La actividad reducida de KMO y la expresión de ARNm de KMO en cerebros esquizofrénicos post-mortem [73] son consistentes con un exceso de respuesta Th2 (Figura 1). Aumento de la prevalencia de anomalías de la inmunidad humoral mediada por Th2 en subgrupos de pacientes con esquizofrenia, como lo demuestra el aumento de los recuentos de células B [21,76], el aumento la producción de autoanticuerpos, incluidos los anticuerpos antivirales [76] y el aumento de la inmunoglobulina E [52], agrega más apoyo a la hipótesis de dominancia de la respuesta Th2.
Neuroinflamación y desregulación del glutamato del SNC
El glutamato media la cognición y el comportamiento [186]. Los niveles séricos de glutamato están regulados por EAAT glial y neuronal dependientes de sodio de alta afinidad, es decir, el sistema XAG responsable de la liberación de glutamato / aspartato [137,164] y el sistema antiportador astrágalo-glutamato / cistina independiente del sodio (Xc-) responsable de la liberación de glutamato / recaptación de cistina [164]. Astroglial EAAT1 y EAAT2 proporcionan más del 90% de recaptación de glutamato [79].
La neuroinflamación puede alterar el metabolismo del glutamato y la función de sus transportadores [15,29,187,188], produciendo alteraciones cognitivas, conductuales y psiquiátricas [15,21,29,79,186,188,189]. Las anormalidades de la función / expresión de EAAT y el metabolismo de glutamato en MDD, BPD, esquizofrenia y TOC se resumen en la Tabla 2.
En MDD, hay evidencia de hiperglutamatergia cortical (Tabla 2). Los niveles de glutamato cortical se correlacionaron positivamente con la gravedad de los síntomas depresivos, y un ciclo de antidepresivos de cinco semanas disminuyó las concentraciones séricas de glutamato [85,86]. Una sola dosis de ketamina, un potente antagonista de NMDAR, puede revertir MDD refractario durante una semana [17,21,29,85]. Los niveles de glutamato en el SNC en exceso pueden inducir inflamación mediada por neurotoxicidad [163,164,188], incluida una respuesta proinflamatoria de Th1 (Figura 2) [164].
Evidencia in vitro limitada sugiere que las citocinas inflamatorias/proinflamatorias pueden aumentar los niveles de glutamato en el SNC [188] en un ciclo de avance a través de varios mecanismos potenciales: 1) las citocinas proinflamatorias pueden inhibir [15,17,168] y revertir [45,137] el glutamato mediado por EAAT astroglial función de recaptación; 2) las citoquinas proinflamatorias pueden mejorar la síntesis de ácido quinolínico microglial [53], que se ha demostrado experimentalmente que promueve la liberación de glutamato sinaptosómico [15,17,29,190]; 3) el aumento de los niveles de COX-2/PGE-2 y TNF-α puede inducir la entrada de calcio [137], que, según los datos in vitro, puede aumentar la liberación de glutamato astroglial y D-serina [191]; y 4) la microglía activada puede expresar un exceso de sistemas antiportadores Xc que median la liberación de glutamato [164,192].
En la esquizofrenia se encuentra hipoglutamatergia cortical prefrontal [87,90,193,194] (Tabla 2) y funcionalidad reducida del NMDAR [5]. Un metanálisis reciente de espectroscopía de resonancia magnética (MRS) H1 (28 estudios, 647 esquizofrenia, 608 control) confirmó una disminución del glutamato y un aumento de los niveles de glutamina en la corteza frontal medial [90]. El papel contribuyente de la inflamación a la hipoglutamatergia no está probado. La síntesis elevada de KYNA en cerebros con esquizofrenia [21,52], típicamente una función de la respuesta Th2 (Figura 1), puede inhibir la subunidad NR1 de NMDAR y alfa 7 nicotínico. receptor de acetilcolina (α7nAchR) [195], lo que conduce a una disminución de la función NMDAR y una reducción de la liberación de glutamato mediada por α7nAchR [195].
En el TLP y el TOC, los datos sugieren hiperglutamatergia cortical del SNC en ambos trastornos (Tabla 2) [78,84,88,131]. La contribución de la inflamación (DBP y TOC) y de los autoanticuerpos (TOC) [7,77,84,88,130] al aumento de los niveles de glutamato en el SNC requiere una mayor investigación.
El papel de S100B
S100B es una proteína de unión a calcio de 10 kDa producida por astroglía, oligodendroglía y células ependimales del plexo coroideo [196]. Media sus efectos sobre las neuronas circundantes y la glía a través del receptor para el producto final de glicación avanzada [196]. Los niveles extracelulares nanomolares de S100B proporcionan efectos neurotróficos beneficiosos, limitan la lesión neuronal relacionada con el estrés, inhiben la liberación de TNF-α microglial y aumentan la recaptación de glutamato astroglial [196]. Las concentraciones micromolares de S100B, predominantemente producidas por astroglía y linfocitos activados [196,197], tienen efectos nocivos transducidos por el receptor para el producto final de glicación avanzada que incluyen apoptosis neuronal, producción de COX-2/PGE-2, IL-1β y especies de óxido nítrico inducible, y regulación positiva de la secreción de TNF-α monocítica/microglial [21,196,198].
Suero y, particularmente, CSF y tejido cerebral Los niveles S100B son indicadores de la activación glial (predominantemente astroglial) [199]. En el TDM y la psicosis, los niveles séricos de S100B se correlacionan positivamente con la gravedad de la tendencia suicida, independientemente del diagnóstico psiquiátrico [200]. El análisis post-mortem de S100B mostró niveles disminuidos en la corteza prefrontal dorsolateral de MDD y BPD, y niveles aumentados en la corteza parietal de BPD [196].
El metanálisis (trastorno del estado de ánimo 193, controles sanos 132) confirmó los niveles elevados de suero y CSF S100B en los trastornos del estado de ánimo, particularmente durante los episodios depresivos agudos y la manía [201].
En la esquizofrenia, los niveles de S100B en el cerebro, el LCR y el suero están elevados [199,202]. El metanálisis (12 estudios, 380 esquizofrenia, 358 controles sanos) confirmó niveles séricos elevados de S100B en la esquizofrenia [203]. En cerebros post mortem de sujetos con esquizofrenia, se encuentran astroglías inmunorreactivas S100B en áreas implicadas en la esquizofrenia, incluida la corteza cingulada anterior, la corteza prefrontal dorsolateral, la corteza orbitofrontal y el hipocampo [154]. Los niveles elevados de S100B se correlacionan con psicosis paranoide [154] y negativista [204], deterioro cognitivo, respuesta terapéutica deficiente y duración de la enfermedad [202]. Los polimorfismos genéticos en S100B [32] y los genes del receptor para el producto final de la glicación avanzada en cohortes de esquizofrenia (Tabla 2) [32,33,205] sugieren que estas anomalías probablemente sean primarias/patógenas en lugar de secundarias/biomarcadores. De hecho, la disminución de los niveles séricos de S100B después del tratamiento con antidepresivos [201] y antipsicóticos [196] sugiere cierta relevancia clínica de S100B para la fisiopatología de los trastornos psiquiátricos.
Neuroinflamación y aumento del estrés oxidativo
El estrés oxidativo es una condición en la cual un exceso de oxidantes daña o modifica las macromoléculas biológicas tales como lípidos, proteínas y ADN [206-209]. Este exceso resulta de una mayor producción de oxidantes, disminución de la eliminación de oxidantes, defensas antioxidantes defectuosas o alguna combinación de los mismos [206-209]. El cerebro es particularmente vulnerable al estrés oxidativo debido a: 1) cantidades elevadas de ácidos grasos poliinsaturados peroxicables; 2) contenido relativamente alto de minerales traza que inducen peroxidación lipídica y radicales de oxígeno (por ejemplo, hierro, cobre); 3) alta utilización de oxígeno; y 3) mecanismos de oxidación limitados [206,207].
El exceso de estrés oxidativo puede ocurrir en MDD [206], BPD [206,207], esquizofrenia [207,209] y TOC [206,208]. Los marcadores periféricos de alteraciones oxidativas incluyen productos de peroxidación lipídica aumentada (por ejemplo, malondialdehído y 4-hidroxi-2-nonenal), metabolitos incrementados de óxido nítrico (NO), antioxidantes disminuidos (por ejemplo, glutatión) y niveles alterados de enzimas antioxidantes [206,207].
En MDD, el aumento de la producción de anión radical superóxido se correlaciona con un aumento de la apoptosis de neutrófilos mediada por oxidación [206]. Los niveles séricos de enzimas antioxidantes (por ejemplo, superóxido dismutasa-1) se elevan durante los episodios depresivos agudos y se normalizan después del tratamiento con inhibidores selectivos de la recaptación de serotonina (ISRS) [206]. Esto sugiere que en MDD, los niveles de enzimas antioxidantes en suero son un marcador de estado, lo que puede reflejar un mecanismo compensatorio que contrarresta los aumentos agudos en el estrés oxidativo. [206]. Por el contrario, en la esquizofrenia, los niveles de superóxido dismutasa-1 solubles en LCR disminuyen sustancialmente en pacientes esquizofrénicos de inicio temprano en relación con pacientes esquizofrénicos crónicos y controles sanos. Esto sugiere que niveles reducidos de enzimas antioxidantes en el cerebro pueden contribuir al daño oxidativo en la esquizofrenia aguda [210], aunque se necesitan estudios más amplios para confirmar este hallazgo.
Varios estudios experimentales y humanos adicionales examinaron con más detalle los mecanismos subyacentes a la fisiopatología del aumento del estrés oxidativo en trastornos psiquiátricos [206-262]. En modelos animales de depresión, los niveles cerebrales de glutatión se reducen, mientras que la peroxidación lipídica y los niveles de NO aumentan [206,262].
Los estudios post mortem muestran niveles cerebrales reducidos de glutatión total en MDD, BPD [206] y sujetos esquizofrénicos [206,207]. Los fibroblastos cultivados de pacientes con MDD muestran un aumento del estrés oxidativo independiente de los niveles de glutatión [262], argumentando contra un papel principal del agotamiento del glutatión como el principal mecanismo del estrés oxidativo en la depresión.
La activación microglial puede aumentar el estrés oxidativo a través de su producción de citocinas proinflamatorias y NO [206-209]. Las citoquinas proinflamatorias y los altos niveles de NO pueden promover la formación de especies reactivas de oxígeno (ROS), lo que a su vez acelera la peroxidación de lípidos, dañando los fosfolípidos de la membrana y sus receptores de neurotransmisores de monoamina unidos a la membrana y agotando los antioxidantes endógenos. El aumento de los productos de ROS puede mejorar la activación microglial y aumentar la producción proinflamatoria a través de la estimulación de NF-κB [208], que a su vez perpetúa la lesión oxidativa [208], creando el potencial para un ciclo de retroalimentación positiva patológica en algunos trastornos psiquiátricos [206-209]. Aunque la neuroinflamación puede aumentar los niveles de glutamato en el cerebro [85,86], el papel de la hiperactividad glutamatérgica como causa del estrés oxidativo sigue sin demostrarse [207].
La disfunción mitocondrial puede contribuir al aumento del estrés oxidativo en MDD, BPD y esquizofrenia [206]. Los estudios post mórtem en estos trastornos revelan anomalías en el ADN mitocondrial, en consonancia con la alta prevalencia de trastornos psiquiátricos en los trastornos mitocondriales primarios [206]. Los estudios en animales in vitro muestran que las citocinas proinflamatorias, como TNF-α, pueden reducir la densidad mitocondrial y alterar el metabolismo oxidativo mitocondrial [211,212], lo que lleva a una mayor producción de ROS [206,213]. Estos hallazgos experimentales pueden implicar vínculos mecánicos entre la neuroinflamación, la disfunción mitocondrial y el estrés oxidativo [206,213], lo que amerita una mayor investigación de estas vías patogénicas que se cruzan en los trastornos psiquiátricos humanos.
La vulnerabilidad del tejido neural al daño oxidativo varía entre diferentes trastornos psiquiátricos basados en las vías neuroanatómicas, neuroquímicas y moleculares involucradas en el trastorno específico [207]. Los efectos del tratamiento también pueden ser críticos, ya que la evidencia preliminar sugiere que los antipsicóticos, los ISRS y los estabilizadores del estado de ánimo poseen propiedades antioxidantes [206,207,262]. El papel terapéutico de los antioxidantes adyuvantes (por ejemplo, las vitaminas C y E) en el trastorno psiquiátrico aún no se ha confirmado mediante ensayos clínicos aleatorizados de gran potencia. La N-acetilcisteína muestra los resultados más prometedores hasta la fecha, con varios ensayos aleatorizados controlados con placebo que demuestran su eficacia en el TDM, la DBP y la esquizofrenia [207].
Disfunción de la barrera hematoencefálica
El BBB asegura el estado inmunológico privilegiado del cerebro al restringir la entrada de mediadores inflamatorios periféricos, incluidas las citocinas y los anticuerpos que pueden afectar la neurotransmisión [214,215]. La hipótesis del colapso de la BBB y su papel en algunos pacientes psiquiátricos [60,214,216,217] es consistente con la mayor prevalencia de comorbilidad psiquiátrica en enfermedades asociadas con su disfunción, incluido el LES [97], el accidente cerebrovascular [11], epilepsia [218] y encefalitis autoinmunes (Tabla 1). Una 'relación LCR:albúmina sérica' elevada en pacientes con MDD y esquizofrenia sugiere una mayor permeabilidad de la BHE [214].
En un estudio (sujetos psiquiátricos 63, controles 4,100), se detectaron anomalías de LCR indicativas de daño de BBB en 41% de sujetos psiquiátricos (14 MDD y BPD, esquizofrenia 14), incluida la síntesis intratecal de IgG, IgM y / o IgA, pleocitosis leve del LCR (células 5 a 8 por mm3) y la presencia de hasta cuatro bandas oligoclonales IgG [216]. Un estudio ultraestructural post mortem en la esquizofrenia reveló anormalidades ultraestructurales de la BBB en las cortezas prefrontal y visual, que incluían la degeneración vacuolar de las células endoteliales, los procesos del pie extremo astroglial y el engrosamiento e irregularidad de la lámina basal [60]. Sin embargo, en este estudio, los autores no comentaron sobre la posible contribución de los cambios postmortem a sus hallazgos. Otro estudio que investiga la transcriptómica de las células endoteliales de BBB en cerebros esquizofrénicos identificó diferencias significativas entre los genes que influyen en la función inmunológica, que no se detectaron en los controles [217].
La disfunción endotelial mediada por la oxidación puede contribuir a la fisiopatología de la disfunción de la BHE en los trastornos psiquiátricos. La evidencia indirecta de los estudios clínicos y experimentales en la depresión [219] y, en menor medida, en la esquizofrenia [220] sugiere que el aumento de la oxidación puede contribuir a la disfunción endotelial. La disfunción endotelial puede representar un mecanismo compartido que representa la asociación conocida entre depresión y enfermedad cardiovascular [219,221], que puede estar relacionada con niveles disminuidos de NO vasodilatador [221-223]. Los estudios experimentales sugieren que los niveles reducidos de NO endotelial se relacionan mecánicamente con el desacoplamiento de la óxido nítrico sintetasa endotelial (eNOS) de su co-factor esencial tetrahidrobiopterina (BH4), desplazando su sustrato de L-arginina a oxígeno [224-226]. La eNOS desacoplada promueve la síntesis de ROS (por ejemplo, superóxido) y especies reactivas del nitrógeno (RNS) (por ejemplo, peroxinitrito, un producto de la interacción de superóxido con NO) [227] en lugar de NO, lo que conduce a una disfunción endotelial mediada por oxidación [ 224-226].
Los datos en animales mostraron que los ISRS podrían restablecer niveles deficientes de NO endotelial [219], lo que sugiere que los mecanismos antioxidantes pueden contribuir a sus efectos antidepresivos. En humanos, el L-metilfolato puede potenciar los efectos antidepresivos de los ISRS [228], supuestamente al aumentar los niveles de BH4, que es un cofactor esencial para la anti-oxidación mediada por reacoplamiento de eNOS [229], así como también para la tasa -limitación de enzimas de síntesis de monoamina (es decir, serotonina, norepinefrina, dopamina) [228].
En conjunto, tanto el trabajo reciente que enfatiza el papel del estrés oxidativo inducido por eNOS desacoplado en la patogenia de las enfermedades vasculares [230,231] como el los estudios epidemiológicos que establecen que la depresión es un factor de riesgo independiente para las patologías vasculares, como el accidente cerebrovascular y la enfermedad cardíaca [219,221], añaden más apoyo a la relevancia clínica del daño oxidativo endotelial no acoplado eNOS en la depresión. A pesar de la abundante evidencia de anomalías de citocinas en enfermedades psiquiátricas humanas y los datos experimentales que muestran que las citoquinas proinflamatorias pueden reducir la expresión de eNOS [212] y aumentar la permeabilidad de BBB [215], la evidencia humana que vincula directamente el exceso de citoquinas proinflamatorias a la disfunción eNOS y / o carente.
Imágenes y tratamiento de la inflamación en enfermedades psiquiátricas
Imaging Neuroinflammation In Situ
Clínicamente, las imágenes de neuroinflamación pueden resultar cruciales para identificar el subgrupo de pacientes psiquiátricos con neuroinflamación que sería más probable que respondan favorablemente a las terapias inmunomoduladoras. Además, tales imágenes pueden permitir a los médicos controlar la actividad de la enfermedad relacionada con la neuroinflamación y su respuesta a la terapia inmune en pacientes psiquiátricos. La inflamación de imágenes en el cerebro humano ha dependido tradicionalmente de la visualización por MRI o TC de los agentes de contraste intravenosos extravagantes, lo que indica una degradación localizada de la BHE. La resonancia magnética mejorada con gadolinio ocasionalmente demuestra dicha descomposición en las regiones límbicas asociadas con el procesamiento emocional en pacientes con trastornos psiquiátricos atribuibles a paraneoplasias u otras encefalitis [107,109,113]. Hasta donde sabemos, sin embargo, nunca se ha demostrado una mejoría anormal en ningún trastorno psiquiátrico clásico [21,214,232], a pesar de las anormalidades funcionales [214,216] y ultraestructurales de la BBB [60].
Aún se desconoce si la neuroinflamación más sutil puede visualizarse in vivo en trastornos psiquiátricos clásicos. Una técnica prometedora es la tomografía por emisión de positrones (PET) utilizando radiotrazadores, como C11-PK11195, que se unen a la proteína translocadora, conocida anteriormente como receptor periférico de benzodiazepinas, expresada por microglia activada [233,234].
Usando este método, los pacientes con esquizofrenia mostraron una mayor activación microglial a lo largo de la corteza [235] y en el hipocampo durante la psicosis aguda [236]. Un estudio (esquizofrenia 14, controles 14) no encontró diferencias significativas entre la unión de [11C] DAA1106 en la esquizofrenia frente a los controles, pero una correlación directa entre la unión de [11C] DAA1106 y la severidad de los síntomas positivos y la duración de la enfermedad en la esquizofrenia [236].
Investigadores de nuestra institución utilizaron C11-PK11195 PET para demostrar inflamación bihipocámpica en un paciente con disfunción neuropsiquiátrica, que incluye TDM psicótico, epilepsia y amnesia anterógrada, asociada con anticuerpos anti-GAD [237]. Sin embargo, PK11195 PET tiene bajas propiedades de señal a ruido y requiere un ciclotrón en el sitio.
En consecuencia, la investigación se está dedicando al desarrollo de ligandos de proteína translocadora mejorados para PET y SPECT. Se necesitan futuros estudios de tejidos cerebrales post-mortem de gran potencia que utilicen la cuantificación de proteínas para dilucidar las vías metabólicas e inflamatorias, las citocinas del SNC y sus receptores de unión en los trastornos psiquiátricos para avanzar en nuestra comprensión de la fisiopatología autoinmune.
Papel de los medicamentos antiinflamatorios en los trastornos psiquiátricos
Varios estudios en humanos y en animales sugieren que ciertos fármacos antiinflamatorios pueden desempeñar un papel coadyuvante importante en el tratamiento de los trastornos psiquiátricos (Tabla 3). Los fármacos comunes son los inhibidores de la ciclooxigenasa (Tabla 3) [238-245], la minociclina (Tabla 3) [240-245], los ácidos grasos omega-3 [246,247] y los neuroesteroides [248].
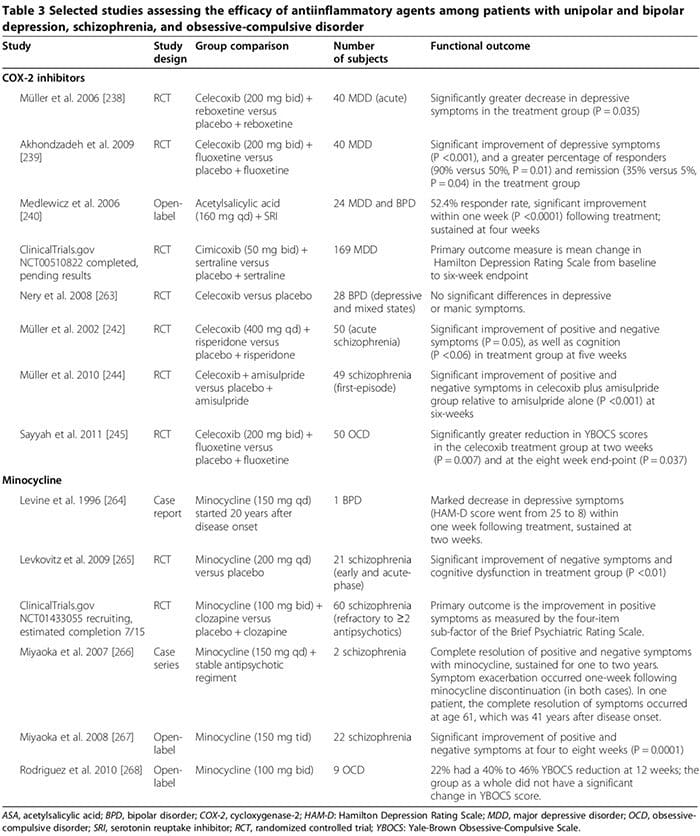 Varios estudios en humanos mostraron que los inhibidores de COX-2 podrían mejorar los síntomas psiquiátricos de MDD, BPD, esquizofrenia y TOC (Tabla 3) [248]. Por el contrario, el tratamiento adyuvante con inhibidores COX no selectivos (es decir, fármacos antiinflamatorios no esteroideos (AINE)) puede reducir la eficacia de los ISRS [249,250]; dos ensayos grandes informaron que la exposición a los AINE (pero no a los inhibidores selectivos de la COX-2 o a los salicilatos) se asoció con un empeoramiento significativo de la depresión entre un subconjunto de participantes del estudio [249,250].
Varios estudios en humanos mostraron que los inhibidores de COX-2 podrían mejorar los síntomas psiquiátricos de MDD, BPD, esquizofrenia y TOC (Tabla 3) [248]. Por el contrario, el tratamiento adyuvante con inhibidores COX no selectivos (es decir, fármacos antiinflamatorios no esteroideos (AINE)) puede reducir la eficacia de los ISRS [249,250]; dos ensayos grandes informaron que la exposición a los AINE (pero no a los inhibidores selectivos de la COX-2 o a los salicilatos) se asoció con un empeoramiento significativo de la depresión entre un subconjunto de participantes del estudio [249,250].
En el primer ensayo, con pacientes deprimidos 1,258 tratados con citalopram durante semanas 12, la tasa de remisión fue significativamente menor entre los que habían tomado AINE al menos una vez en comparación con los que no (45% frente a 55%, O 0.64, P = 0.0002) [249]. El otro ensayo, que involucró a sujetos con 1,545 MDD, mostró que la tasa de depresión resistente al tratamiento fue significativamente mayor entre los que tomaban AINE (o 1.55, 95% CI 1.21 a 2.00) [231]. El empeoramiento de la depresión en los grupos de AINE puede no estar relacionado mecánicamente con el tratamiento con AINE sino con afecciones médicas crónicas coexistentes [10,12-18] que requieren AINE a largo plazo y que se sabe que están asociadas de forma independiente con un mayor riesgo de la depresión resistente al tratamiento [249,251]. Se necesitan estudios futuros que investiguen el impacto de los AINE en la depresión y la respuesta a los antidepresivos en humanos.
En otros estudios experimentales que utilizaron paradigmas de estrés agudo para inducir un estado similar a la depresión en ratones, el citalopram aumentó el TNF-α, IFN-γ y p11 (factor molecular relacionado con el comportamiento depresivo en animales) en la corteza frontal, mientras que el AINE el ibuprofeno disminuyó estas moléculas; Los AINE también atenuaron los efectos antidepresivos de los ISRS, pero no de otros antidepresivos [249]. Estos hallazgos sugieren que las citoquinas proinflamatorias pueden, paradójicamente, ejercer efectos antidepresivos a pesar de la abrumadora evidencia de los estudios en humanos indican lo contrario (como se revisó anteriormente), lo que puede atenuarse con los AINE [249]. Al menos dos consideraciones pueden explicar esta aparente paradoja: 1) en algunas condiciones experimentales, las citoquinas proinflamatorias se han asociado con un papel neuroprotector, [251; (para ejemplo, IFN-γ en niveles bajos puede inducir microglía neuroprotectora (Figura 2) [163,166,251]); y 2) aún no está claro si estas respuestas observadas en el contexto de un paradigma de estrés agudo en un modelo animal son aplicables al MDD endógeno en humanos [251].
Los efectos terapéuticos de los inhibidores de la COX-2 en los trastornos psiquiátricos pueden implicar la modulación de la biosíntesis de las prostaglandinas derivadas de la COX-2, incluida la PGE2 proinflamatoria y la 15-desoxi-12,14-PGJ2 antiinflamatoria (15d-PGJ2) [252,253]. Los inhibidores de la COX-2 pueden reducir la inflamación mediada por PGE2, lo que puede contribuir a la fisiopatología de los trastornos psiquiátricos [252,253]. También pueden alterar los niveles de 15d-PGJ2 y la actividad de su receptor nuclear gamma activado por el proliferador de peroxisomas (PPAR-γ) [252,253].
Varios estudios sugieren que 15d-PGJ2 y su receptor nuclear PPAR-γ pueden servir como marcadores biológicos para la esquizofrenia [253]. En pacientes esquizofrénicos, los niveles séricos de PGE2 aumentan, mientras que los niveles séricos de 15d-PGJ2 disminuyen, al igual que la expresión de su receptor nuclear PPAR-γ en PBMC [252]. Si bien los inhibidores de la COX-2 pueden limitar los efectos antiinflamatorios potencialmente beneficiosos de la 'vía 2d-PGJ15/PPAR-γ' dependiente de la COX-2, pueden reducir ventajosamente sus efectos nocivos, incluido 1) el aumento del riesgo para infarto de miocardio y ciertas infecciones (por ejemplo, citomegalovirus y Toxoplasma gondii) en pacientes esquizofrénicos [254] y 2) sus efectos proapoptóticos observados en tejido canceroso humano y animal [255]. Otros posibles mecanismos de los efectos terapéuticos de los inhibidores de la COX-2 pueden implicar su capacidad para reducir los niveles de citocinas proinflamatorias [163], limitar la excitotoxicidad del ácido quinolínico (como en el MDD) y disminuir los niveles de KYNA (como en la esquizofrenia) [128].
La minociclina puede ser eficaz en los trastornos psiquiátricos (tabla 3) [248]. Los datos in vitro sugieren que la minociclina inhibe MAP, la secreción de citocinas, la expresión de COX-2/PGE-2 y la óxido nítrico sintasa inducible [256]. La minociclina también puede contrarrestar la neurotransmisión glutamatérgica y dopaminérgica desregulada [256].
La eficacia de los ácidos grasos omega-3 en los trastornos psiquiátricos no está clara [248]. En un metanálisis de 2011 de 15 ensayos controlados aleatorios (916 MDD), los suplementos de omega-3 que contenían ácido eicosapentaenoico ≥60 % (rango de dosis de 200 a 2,200 mg/d por encima de la dosis de ácido docosahexaenoico) redujeron significativamente los síntomas depresivos como terapia complementaria a los SRI (P <0.001) [246]. Sin embargo, un metanálisis posterior concluyó que los ácidos grasos omega-3 no tienen un beneficio significativo en la depresión y que la supuesta eficacia es simplemente el resultado del sesgo de publicación [247]. Un metanálisis de 2012 de 5 ensayos controlados aleatorios que incluyeron a 291 participantes con TLP encontró que los síntomas depresivos, pero no maníacos, mejoraron significativamente entre los asignados al azar a los ácidos grasos omega-3 en relación con los que tomaron placebo (Hedges g 0.34, P = 0.025) [257]. En un ensayo controlado aleatorizado de sujetos esquizofrénicos seguidos hasta 12 meses, las puntuaciones de los síntomas positivos y negativos se redujeron significativamente entre los 66 participantes aleatorizados a omega-3 de cadena larga (1.2 g/día durante 12 semanas; P = 0.02 y 0.01, respectivamente) [258]; la A Los autores concluyeron que el aumento omega-3 durante el curso temprano de la esquizofrenia también puede prevenir las recaídas y la progresión de la enfermedad [258].
Un metanálisis 2012 de siete ensayos controlados aleatorios que evaluaron el aumento de omega-3 en pacientes esquizofrénicos 168 no encontró ningún beneficio del tratamiento [259]. Los autores de este metanálisis expresaron específicamente que no se podía establecer ninguna conclusión con respecto a la prevención de recaídas o los criterios de valoración de la progresión de la enfermedad [259]. Los datos experimentales sugieren que el ácido eicosapentaenoico y el ácido docosahexaenoico median sus efectos antiinflamatorios promoviendo la síntesis de resolvinas y protectinas, que pueden inhibir la infiltración de leucocitos y reducir la producción de citocinas [248].
Los neuroesteroides, incluida la pregnenolona y su alopregnanolona de metabolito descendente, pueden tener un papel beneficioso en algunos trastornos psiquiátricos [248,260]. En el TDM, varios estudios encontraron una disminución de los niveles de alopregnanolona en plasma / LCR que se correlacionaba con la gravedad de los síntomas, que se normalizó después de un tratamiento exitoso con ciertos antidepresivos (por ejemplo, ISRS) y terapia electroconvulsiva [261]. En la esquizofrenia, los niveles de pregnenolona en el cerebro pueden verse alterados [248] y los niveles de alopregnanolona sérica pueden aumentar después de algunos medicamentos antipsicóticos (por ejemplo, clozapina y olanzapina) [260]. En tres ensayos controlados aleatorios (esquizofrenia 100 (agrupados), duración del tratamiento, aproximadamente nueve semanas), los síntomas positivos, negativos y cognitivos, así como los efectos secundarios extrapiramidales de los antipsicóticos mejoraron significativamente en uno o más ensayos entre los aleatorizados a pregnenolona en relación con los que recibieron placebo [248]. En un ensayo, la mejoría se mantuvo con el tratamiento con pregnenolona a largo plazo [248]. La pregnenolona puede regular la cognición y el comportamiento al potenciar la función de los receptores NMDA y GABAA [248]. Además, la alopregnanolona puede ejercer efectos neuroprotectores y antiinflamatorios [248]. Se necesitan más estudios ECA para confirmar la función beneficiosa de los esteroides neuroactivos en los trastornos psiquiátricos de aparición temprana en los seres humanos.
Estamos a la espera de los resultados de varios ensayos clínicos en curso que investigan los efectos terapéuticos de otros agentes antiinflamatorios, incluido el salicilato, un inhibidor de NF-κB (NCT01182727); ácido acetilsalicílico (NCT01320982); pravastatina (NCT1082588); y dextrometorfano, un antagonista de NMDAR no competitivo que puede limitar la lesión neuronal dopaminérgica inducida por inflamación (NCT01189006).
Futuras estrategias de tratamiento
Aunque las terapias inmunológicas actuales (por ejemplo, IVIG, plasmaféresis, corticosteroides y agentes inmunosupresores) suelen ser eficaces para tratar las encefalitis autoinmunes en las que la inflamación es aguda, intensa y predominantemente de origen adaptativo, su eficacia en los trastornos psiquiátricos clásicos en los que la inflamación es crónica, mucho más leve, y predominantemente de origen innato, es limitado [2]. El desarrollo de nuevas terapias debe apuntar a revertir la pérdida glial [46,138], regulando negativamente el MAP dañino, mientras optimiza los marcadores neuroprotectores endógenos y el MAP beneficioso, en lugar de suprimir indiscriminadamente la inflamación como ocurre con los agentes inmunosupresores actuales. Además, se necesita el desarrollo de potentes antioxidantes coadyuvantes que revertirían la lesión oxidativa en los trastornos psiquiátricos.
Conclusiones
La autoinmunidad puede causar una serie de trastornos neuropsiquiátricos que pueden presentarse inicialmente con síntomas psiquiátricos aislados. La inflamación / autoinmunidad innata puede ser relevante para la patogénesis de los síntomas psiquiátricos en un subconjunto de pacientes con trastornos psiquiátricos clásicos. La inflamación innata puede estar relacionada mecánicamente con las anomalías monoaminérgicas y glutamatérgicas tradicionales y el aumento de la lesión oxidativa informada en enfermedades psiquiátricas.
Souhel Najjar1,5 *, Daniel M Pearlman2,5, Kenneth Alper4, Amanda Najjar3 y Orrin Devinsky1,4,5
Abreviaturas
3-OH-KYN: 3-hidroxi-quinurenina; α7nAchR: receptores nicotínicos de acetilcolina alfa 7; AMPAR: receptores de ácido amino-3-hidroxi-5-metil-4-XNUMX-isoxazolpropiónico; APC: célula presentadora de antígeno; BBB: Barrera hematoencefálica;
BH4: Tetrahidrobiopterina; BPD: trastorno bipolar; IC: intervalo de confianza;
SNC: sistema nervioso central; COX-2: ciclooxegenasa-2; LCR: líquido cefalorraquídeo; DSM-IV: Manual Diagnóstico y Estadístico de los Trastornos Mentales 4th Edition; EAAT: transportadores de aminoácidos excitadores; eNOS: óxido nítrico sintetasa endotelial; GABAB: ácido gamma aminobutírico beta; GAD: ácido glutámico descarboxilasa; GFAP: proteína ácida fibrilar Glial; GLX: 1H MRS glutamato detectable, glutamina, compuesto de ácido gamma aminobutírico;
IDO: Indolamina 2,3-dioxigenasa; Ig: Inmunoglobulina; IL: interleucina; IL-1RA: antagonista del receptor de interleucina 1; IFN-γ: Interferón gamma;
KAT: Kynurenine aminotransferase; KMO: Kynurenine 3-monooxygenase; KYN: Kyurenita; KYNA: ácido Kynurenic; LE: encefalitis límbica;
LPS: lipopolisacárido; MAP: activación y proliferación microglial;
TDM: trastorno depresivo mayor; mGluR: receptor metabotrópico de glutamato; MHC: II Complejo mayor de histocompatibilidad clase dos; MRI: Imágenes por resonancia magnética; MRS: Espectroscopía de resonancia magnética; NF-κB: factor nuclear kappa B; NMDAR: receptor de N-metil-D-aspartato; NR1: sitio de glicina;
TOC: trastorno obsesivo-compulsivo; O: odds ratio; PANDAS: trastornos autoinmunes neuropsiquiátricos pediátricos asociados con infecciones por estreptococos; CMSP: células mononucleares de sangre periférica; PET: tomografía por emisión de positrones; PFC: Corteza prefrontal; PGE-2: prostaglandina E2; PPAR-
γ: receptor nuclear activado por proliferador de peroxisomas gamma; QA: ácido quinolínico; RNS: Especies nitrogenadas reactivas; ROS: especies reactivas de oxígeno;
sIL: interleucina soluble; LES: lupus eritematoso sistémico; SRI: Inhibidor de la recaptación de serotonina; TNF-α: factor de necrosis tumoral alfa; T-regs: células T reguladoras CD4+CD25+FOXP3+; TDO: triptófano-2,3-dioxigenasa; Th: T-ayudante; VGKC: canal de potasio dependiente de voltaje; XAG-: transportador de aspartato de glutamato; Xc-: Glutamato/cistina astroglial independiente de sodio
sistema antiportero
Conflicto de intereses
Los autores declaran que no tienen intereses en conflicto.
Contribuciones de los autores
SN y DMP realizaron una extensa revisión de la literatura, interpretaron los datos, prepararon el manuscrito, las figuras y las tablas. KA preparó la sección relativa a los mecanismos oxidativos y contribuyó a las revisiones del manuscrito. AN y OD revisaron críticamente y mejoraron el diseño y la calidad del manuscrito. Todos los autores leyeron y aprobaron el manuscrito final.
AGRADECIMIENTOS
Agradecemos a los Dres. Josep Dalmau, MD, PhD, Tracy Butler, MD, y David Zazag, MD, PhD, por proporcionar su experiencia en encefalitis autoinmunes, neuroinflamación de imágenes y neuropatología, respectivamente.
Detalles del autor
1Department of Neurology, Facultad de Medicina de la Universidad de Nueva York, 550 First Avenue, Nueva York, NY 10016, EE. UU. 2Geisel School of Medicine en Dartmouth, The Dartmouth Institute for Health Policy and Clinical Practice, 30 Lafayette Street, HB 7252, Líbano, NH 03766, EE. UU. 3Departamento de Patología, División de Neuropatología, Facultad de Medicina de la Universidad de Nueva York, 550 First Avenue, Nueva York, NY 10016, EE. UU. 4Departamento de Psiquiatría, Facultad de Medicina de la Universidad de Nueva York, Nueva York, NY, EE. UU. 5 Centro de Epilepsia Completa de la Universidad de Nueva York, 550 First Avenue, Nueva York, NY 10016, EE. UU.
Publicar descargos de responsabilidad
Alcance de la práctica profesional *
La información aquí contenida en "Neuroinflamación y enfermedad psiquiátrica" no pretende reemplazar una relación personal con un profesional de la salud calificado o un médico con licencia y no es un consejo médico. Lo alentamos a que tome decisiones de atención médica basadas en su investigación y asociación con un profesional de la salud calificado.
Información del blog y debates sobre el alcance
Nuestro alcance informativo se limita a la quiropráctica, musculoesquelética, medicina física, bienestar, contribuyendo etiológico alteraciones viscerosomáticas dentro de las presentaciones clínicas, la dinámica clínica del reflejo somatovisceral asociado, los complejos de subluxación, los problemas de salud delicados y/o los artículos, temas y debates de medicina funcional.
Brindamos y presentamos colaboración clínica con especialistas de diversas disciplinas. Cada especialista se rige por su ámbito de práctica profesional y su jurisdicción de licencia. Utilizamos protocolos funcionales de salud y bienestar para tratar y apoyar la atención de lesiones o trastornos del sistema musculoesquelético.
Nuestros videos, publicaciones, temas, asuntos e ideas cubren cuestiones clínicas, problemas y temas que se relacionan y respaldan directa o indirectamente nuestro ámbito de práctica clínica.*
Nuestra oficina ha intentado razonablemente proporcionar citas de apoyo y ha identificado el estudio o los estudios de investigación relevantes que respaldan nuestras publicaciones. Proporcionamos copias de los estudios de investigación de respaldo disponibles para las juntas reguladoras y el público a pedido.
Entendemos que cubrimos asuntos que requieren una explicación adicional de cómo puede ayudar en un plan de atención o protocolo de tratamiento en particular; por lo tanto, para discutir más a fondo el tema anterior, no dude en preguntar Dr. Alex Jiménez, DC, o póngase en contacto con nosotros en 915-850-0900.
Estamos aquí para ayudarlo a usted y a su familia.
Bendiciones
El Dr. Alex Jimenez corriente continua MSACP, enfermero*, CCCT, IFMCP*, CIFM*, ATN*
email: coach@elpasomedicinafuncional.com
Licenciado como Doctor en Quiropráctica (DC) en Texas & New Mexico*
Número de licencia de Texas DC TX5807, Nuevo México DC Número de licencia NM-DC2182
Licenciada como Enfermera Registrada (RN*) en Florida
Licencia de Florida N.° de licencia de RN RN9617241 (Control No. 3558029)
Estado compacto: Licencia multiestatal: Autorizado para ejercer en 40 Estados*
Matriculado actualmente: ICHS: MSN* FNP (Programa de enfermera practicante familiar)
Dr. Alex Jiménez DC, MSACP, RN* CIFM*, IFMCP*, ATN*, CCST
Mi tarjeta de presentación digital




 De nuevo te doy la bienvenida.
De nuevo te doy la bienvenida.
Los comentarios están cerrados.